论文:Vials: Visualizing Alternative Splicing of Genes
作者:Hendrik Strobelt, Bilal Alsallakh, Joseph Botros, Brant Peterson, Mark Borowsky, Hanspeter Pfister, and Alexander Lex
发表:InfoVis 2015
介绍
在这篇文章中,Hendrik Strobelt等人提出了一种基因选择性剪接的可视化系统。
选择性剪接的工作机制是:同一DNA序列,在转录过程中选择性地省略或缩短一些相关的编码区(外显子),从而生成多种mRNA异构体,指导合成多种多样的蛋白质。
选择性剪接的检测比较困难,需要使用先进的数据采集方法和统计推断方法。目前比较常用的有mRNA测序方法。
关于异构体丰度的研究对于理解正常生理过程和疾病过程具有重要作用,最终可以帮助靶向治疗。由于异构体数据比较复杂,现有的关于异构体的可视化系统既不具备有效的感知性,也不具备可拓展性。为了弥补这些缺点,他们实现了一个可视分析工具,命名为Vials,帮助分析人员探索异构体数据库:包括编码区序列(外显子)的丰度,连接的支持数据,异构体频率的预测等。Vials具有可拓展性,可以支持同时分析多个组内的许多样本。他们的工具可以帮助专家在多组样本中,确定异构体丰度的模式,评估数据的质量等。
数据类型
3种实验数据:
(1) 异构体丰度:
(2) 每个外显子或碱基对的表达数据:
(3) 外显子之间连接支持数据:
2种独立于实验数据的数据:
(1) 参照基因组
(2) 关于外显子和异构体的参考信息: 哪些外显子和异构体是已知存在的
相关工作
Sashimi plots(目前最常用的涉及连接支持和外显子序列的异构体可视化方法):
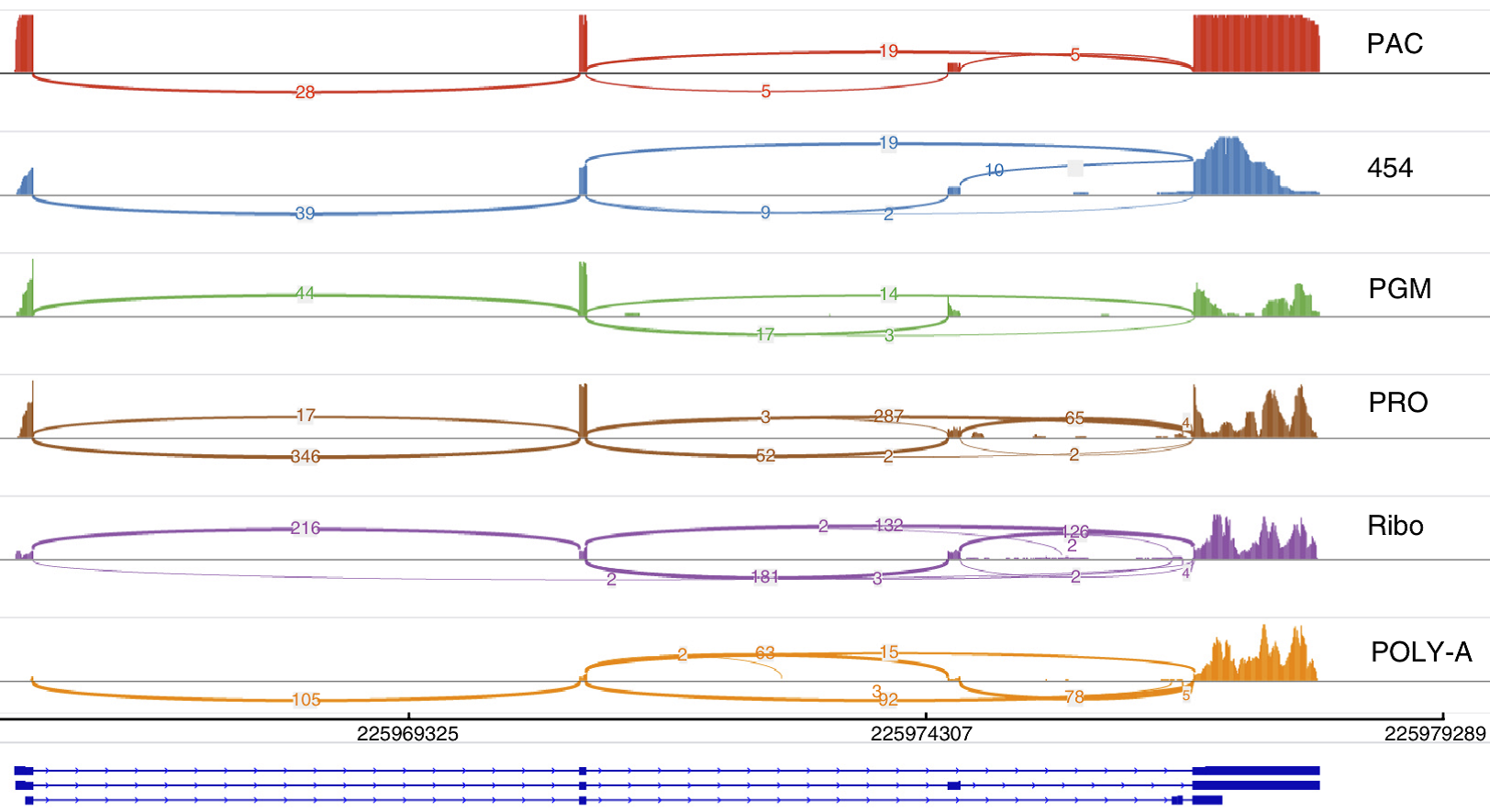
缺点:
用边的宽度来编码连接支持会产生的大范围的数据值并不合适
e.g.第四行中,17和346大约20倍的差距而边的宽度变化不大,所以分析师还是需要依赖边上标记的数值来判断。
不具有可拓展性
e.g.图中展示了一个只有三种异构体的基因,但是可以发现已经存在了很多边的遮挡。一种改进方法是采用交互手段只显示被选择的异构体的连接情况,即本文中采用的方法。
如果碱基对间距比较近,在视觉上也会难以看清。
不支持很多的样本。
任务
关于3种实验数据的任务:
T1: 判断一个样本或样本组的数量级
T2: 比较样本并且确定组内的差异/方差和离群值
T3: 确定组与组之间的差异/方差
关于异构体构成的任务:
C1: 确定外显子/连接是一个异构体的一部分
C2: 确定异构体之间的关系
C3: 确定在已有数据库中不存在的新的外显子或者异构体的存在
系统视图介绍
一共有3种视图
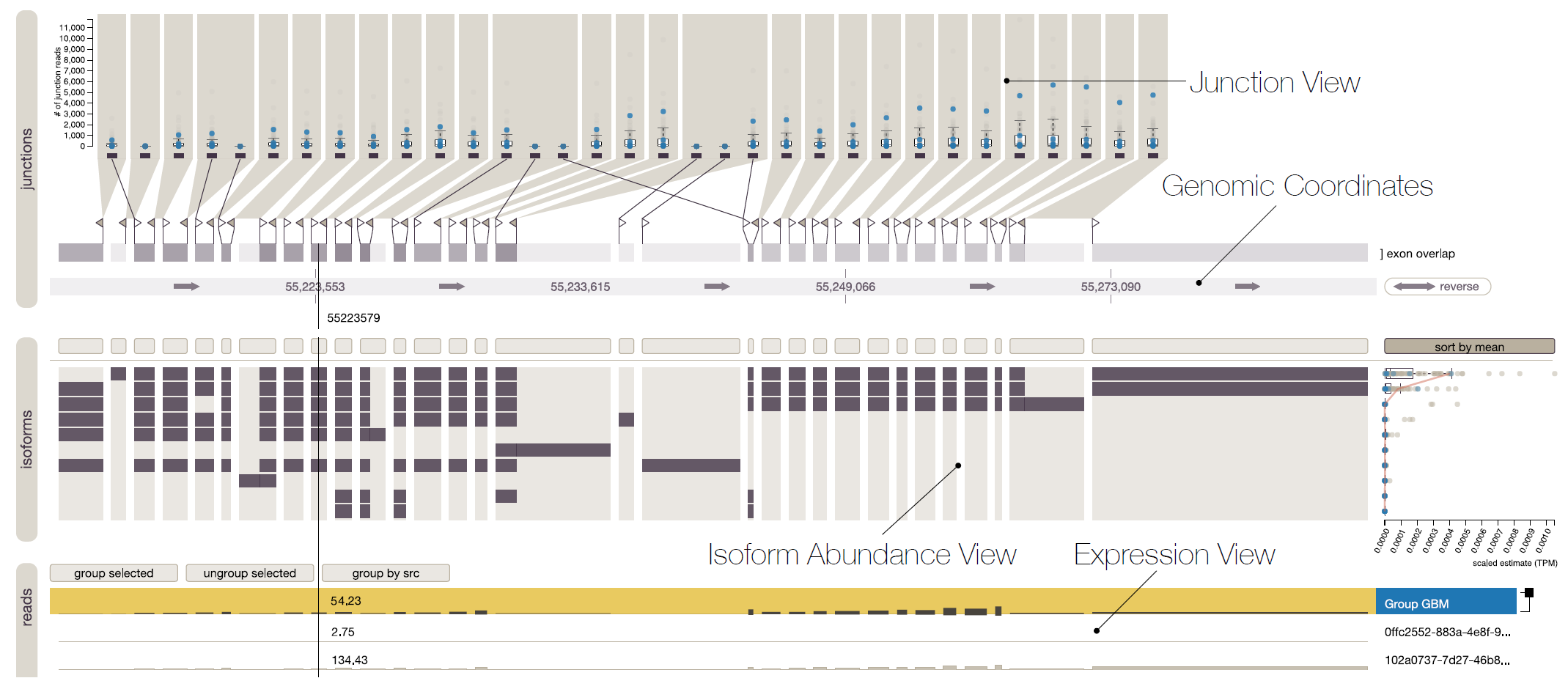
1. Isoform Abundance View 异构体丰度视图

简介:
可视化了数据库中所有的异构体,左侧展示了外显子的结构,右侧用点图和盒须图展示了样本中每一个异构体的丰度值。
可视编码:
黄色边框划分出的区域代表了一个异构体的结构和丰度。左侧的黑色块表示在这个异构体中存在的外显子,浅灰色的区域表示的是至少有一个异构体表达的外显子。
右侧的每一个点表示这个异构体在某一个样本中的丰度值。(他们原本只使用了点图,但是合作者表示在面对大型数据集的时候会有一些混乱,于是增加了盒须图,同时在处理大型数据集时还使用了抖动来分散点,使用了透明度来减少重叠绘制)
交互:
用户可以选择只显示外显子,观察异构体结构;或者包含内含子,观察基因的整体结构。
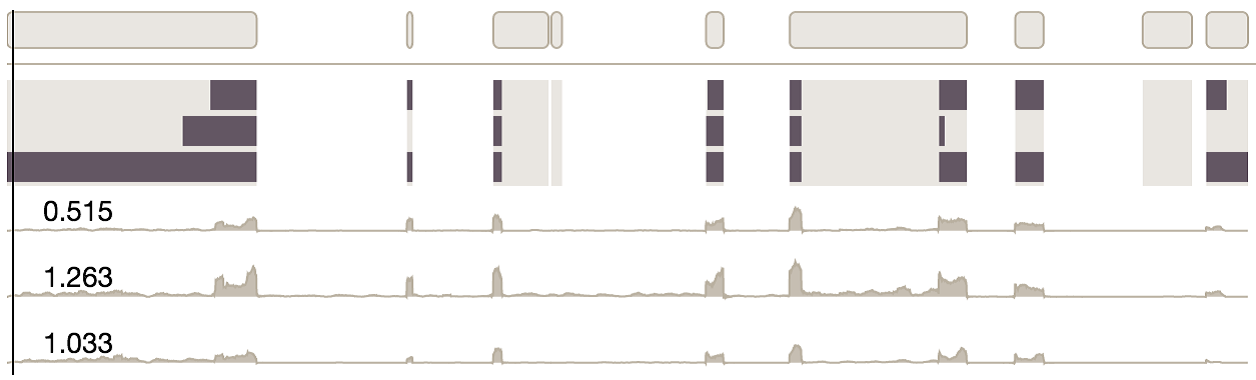
用户可以对异构体进行排序,可根据外显子的包含情况排序,也可以根据外显子丰度的平均值来排序。
用户选中一个样本后,一条带有颜色的线会将这个样本在丰度图中所有的点连接起来。
当一个组被定义了之后,样本点根据不同的组有不同的颜色编码,由于在一个丰度图内会出重叠的情况,所以提供了可以查看细节的交互,在细节视图中,每一个组的样本点都被分开。

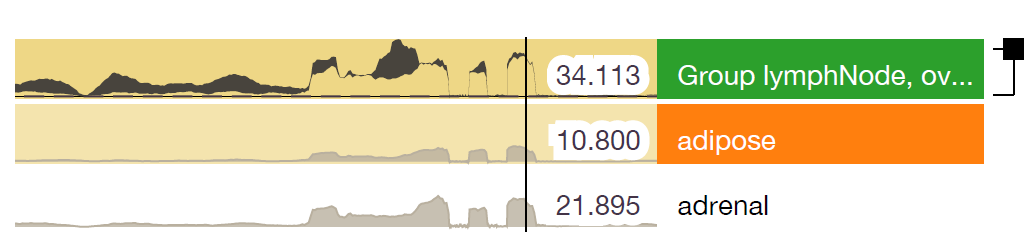
2. Expression View 表达视图
简介:
这个视图展示的是样本和样本组测量出来的外显子丰度分布,视图的位置和基因组坐标轴也是对齐的。可以是per-base-data,也可以是per-exon-data。
可视编码:
用曲线图表示了外显子的丰度分布。
对选择的不同样本和样本组用不同的颜色标记。
交互:
用户在表达视图中进行选择,其他视图中也会有联动。
允许用户定义样本组。
3. Junction View 连接视图
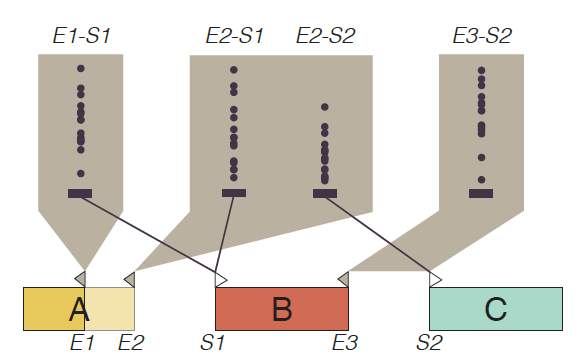
概念:
A、B、C代表了三个外显子,每一个外显子的开始处和结尾处都有标记,在上方绘制一个三角形。从每一个结尾处出发,在上方绘制一个多边形,在多边形里面绘制从这个结尾开始的所有连接,这些连接用点图或盒须图表示。每一个连接都会有一个连线连向另一个外显子开始处,也就是这个连接的结束处。由于有很多连接都是相邻的连接,对于这些相邻的连接,就把多边形的延伸到另一个外显子开始处,把连线省略。
简介:
这个视图展示的是多样本中的连接关系。
视觉编码:
每一个外显子的开始处有一个白色三角形代表连接的结束处。每一个外显子的结束处有一个灰色三角形代表连接的开始处,由灰色三角形延伸出来的多边形中用点图和盒须图表示了所有样本的连接丰度。每一个连接用一条连线连接到连接的结束处,相邻的连接则利用多边形延伸方法省略连线。
交互:
用户可以选择将鼠标移至某一个连接开始处,就会高亮显示该连接开始处的所有连接,省略其他连接。还可以选中某一个异构体,就会高亮显示这个异构体的所有连接,省略其他连接。
还可以将连接视图中的点图和盒须图切换成散点图形式的关系视图。选中一个散点图作为参考散点图,在这个散点图内,所有的样本点递增排列。在其他散点图内,样本点水平方向的位置会根据参考散点图中的位置来排布,因此横坐标相当于参考散点图中连接的数量,所以能反映这些连接和选中连接的关系。

Case Study
1. 癌症样本组中的选择性剪接
他们比较了SRSF7基因在2组样本中的表达区别。第一组是脑癌样本组GBM,一共100个样本,用橙色标记,另一组是白血病样本组LAML,一共167个样本,用蓝色标记。
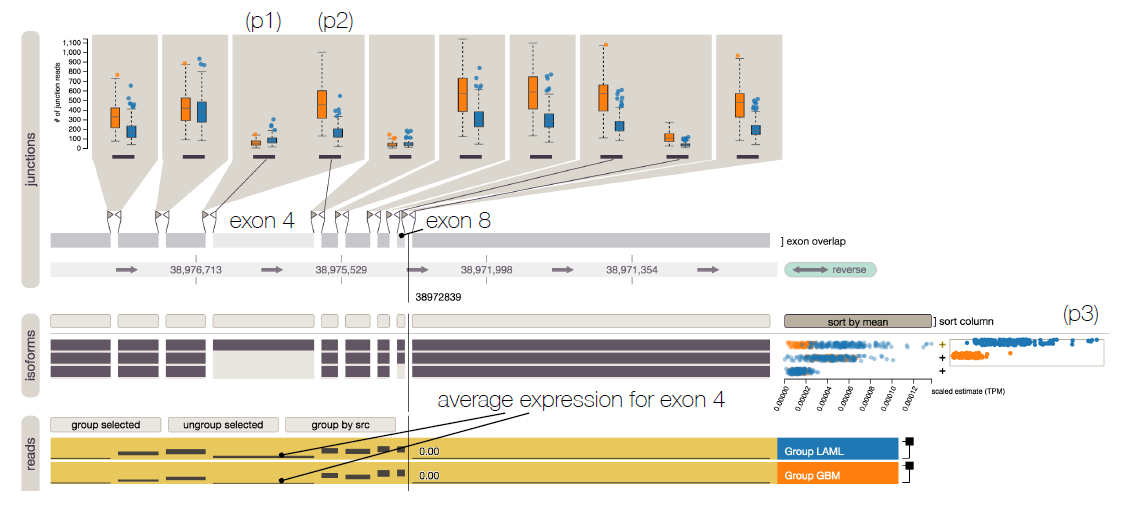
- 1 研究Exon4(外显子4)在LAML和GBM中的使用情况:
首先在表达视图中,可以观察到Exon4在两组中的表达都比较低,相对在LAML中高一点;然后观察到模式p1,发现Exon3和Exon4之间的连接,LAML更多一些;同时观察到模式p2,发现从Exon3开始跳过Exon4的连接,GBM的明显要多;由此说明了Exon4在LAML中的使用确实更多,这在模式p3中也得到了验证:LAML的异构体1的丰度明显高于GBM。
- 2 发现可能的新异构体:
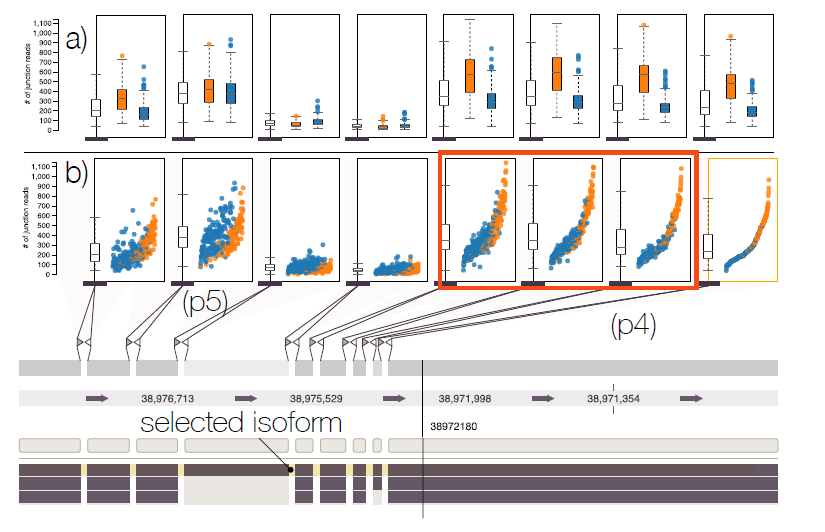
根据Exon8到Exon9之间的连接来重新排布样本点;观察到模式p4,发现在这三个连接处,两个样本表现出来情况的和Exon8-Exon9之间连接是比较相似的;同时观察到模式p5,发现在这个连接处,两个样本集的分散情况明显不同;这表明了有新的异构体存在的可能,可能存在一个没有外显子1和2的新的异构体。
2. 检验数据的质量

他们观察到在丰度视图,白血球样本和其他样本有明显的偏离。所以特地观察了这组样本数据。发现在表达视图,并没有白血球样本相应的表达数据。所以这组数据是有问题的。因此系统可以用来检测数据的质量。
✉️ zjuvis@cad.zju.edu.cn